多氟烷基化合物(PoFAS)是一类结构中含有非全氟基团的全氟化合物(PFAS),在受污染水体及自然环境中可被羟基自由基(•OH,最活跃的活性氧物种之一)氧化并发生转化。然而,不同PoFAS在•OH作用下的反应活性差异、结构对反应位点和路径的影响机制、以及转化产物的具体组成,目前尚不清晰。
近日,西湖大学张岩岩实验室在ENVIRONMENTAL SCIENCE & TECHNOLOGY上发表题为“Structure-dependent reactivity and transformation products ofPolyfluoroalkyl substances by hydroxyl radicals”的研究论文,系统考察了四类共28种PoFAS在UV/H2O2高级氧化体系中的转化行为。该体系可稳定产生•OH、且避免其他活性氧物种的干扰。研究证实,•OH能够将多种非全氟官能团转化为全氟羧酸结构;通过量子化学计算揭示了影响PoFAS反应活性的关键结构特征;并借助高分辨质谱鉴定转化产物,进一步阐明了不同PoFAS的反应路径及关键产物的生成机制。该研究为深入理解新型PoFAS的环境持久性、识别其活性位点及预测转化产物提供了重要理论依据。本研究第一作者是浙江大学、西湖大学联合培养博士研究生单文禹,通讯作者是西湖大学特聘研究员、助理教授张岩岩。

论文链接:https://doi.org/10.1021/acs.est.5c06831
图片摘要
全氟和多氟烷基化合物(
PFAS
)
是
全球高度关注的新污染物
,
在环境中浓度高、难降解、健康危害大,也是氟化工、新能源、消防等领域不可或缺的关键化学品。在产业需求不断攀升、而传统
PFAS
面临全球限制的背景下,为满足多样化功能需求,新型
PFAS
结构不断涌现,尤其是结构中含有非碳氟基团或头基含有烃类的多氟烷基化合物(
PoFAS
)。这些非全氟基团,包括甲基
/
乙基(
−(CH
2
)
n
−
)、氢取代(
−CH
)、烯基(
C=C
)和磺酰胺(
−SO
2
N−
)等,致使
PoFAS
在环境中能够发生非生物或生物转化,最终生成极为稳定的全氟烷基酸(
PFAA
),从而成为
PFAA
重要的间接来源。
羟基自由基(•OH)是自然环境中活性最强的氧化自由基,广泛存在于受太阳辐射的地表水体,同时也是高级氧化水处理工艺中的关键氧化物种。•OH对传统碳氢类污染物表现出优异的降解能力,其二级速率常数(k)达109 M−1·s−1量级。也能够与PoFAS中的非全氟基团反应,将PoFAS转化为末端含羧酸基团的全氟羧酸化合物(PFCA),是PoFAS环境转化行为的重要组成部分。但是,PoFAS在•OH作用下的反应活性、反应位点及转化产物尚不明确。且在实际水体或非理想反应条件下,PoFAS的转化可能停留在仍具有反应活性、结构多样的中间产物阶段,这不仅为环境中PoFAS的赋存形态识别带来分析上的困难,也增加了其潜在生态与健康风险。因此,亟需采用能够稳定产生•OH的可控反应体系(如UV/H2O2),系统研究不同PoFAS的反应动力学及产物组成,这对于准确理解和预测新型PoFAS的环境转化行为具有重要意义。
本研究选取了28种具有代表性的PoFAS(图1a),涵盖污染环境中常见的甲基/乙基类、烯基类、氢取代类和磺酰胺类等典型结构。通过UV/H2O2氧化体系产生•OH,系统测定PoFAS与•OH的二级速率常数,揭示其固有反应活性;进一步结合过渡态理论,阐释导致不同PoFAS反应活性差异的结构因素,包括非全氟基团特征、反应位点、极性基团差异等。基于高分辨质谱鉴定的中间产物,研究阐明了四类PoFAS的转化路径及关键产物的生成机制。该研究为深入理解PoFAS的环境转化行为、评估新型PoFAS的环境持久性、识别易氧化位点、及预测产物组成提供了重要的理论依据。
采用UV/H2O2氧化体系对28种PoFAS进行反应动力学研究,获取其拟一级速率常数,基于•OH稳态浓度([•OH]ss =2.0 × 10⁻12 M)计算相应的二级速率常数(kexp)。结果显示,kexp跨越四个数量级(图1b),充分体现了PoFAS分子结构对其反应活性的显著影响。其中,含双键的PoFAS反应最快,kexp高达0.57−2.02 × 109 M⁻1·s⁻1;其次为含甲基或乙基的PoFAS(0.11−1.57 × 108 M⁻1·s⁻1)和含磺酰胺衍生物的PoFAS(0.40−2.22 × 108 M⁻1·s⁻1);而仅含一个C−H键的H-PoFAS反应最慢,kexp仅为0.47−9.69 × 106 M⁻1·s⁻1。值得注意的是,除6:2 FTSA和NBP2外,其他PoFAS与•OH的二级速率常数均为首次报道。
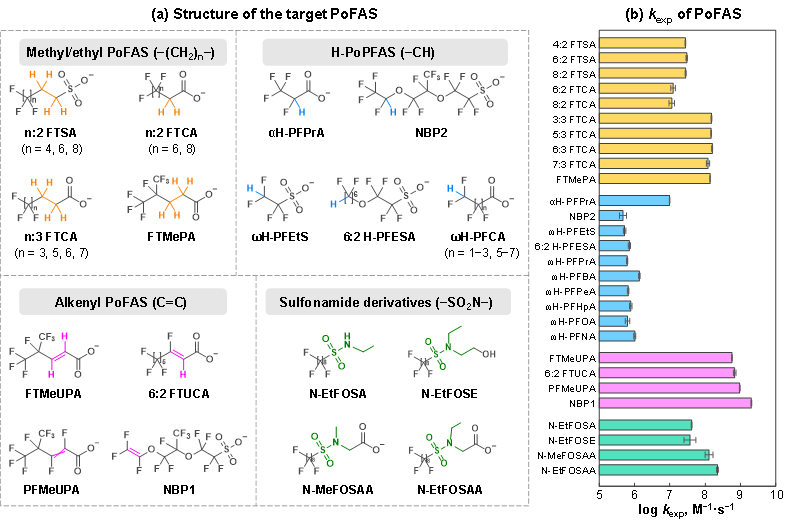
图1. PoFAS结构及其与•OH的二级速率常数(kexp)
•OH主要通过氢提取(H-abstraction)和双键加成两种机制降解污染物,这也是其与PoFAS反应的主要途径。而不同PoFAS反应活性的显著差异,表明了非全氟基团在其转化过程中的关键作用。这些非全氟基团的结构特征、反应位点、极性头基差异、以及空间位阻等,不仅影响反应活性,也直接导致反应路径与最终产物的差异。需综合运用反应动力学分析、转化产物鉴定与理论计算等多种手段,系统阐释非全氟基团影响PoFAS反应活性的内在机制。
以含甲基/乙基的PoFAS为例,其反应活性受结构特征影响显著。含乙基的n:2 FTSA(n = 4, 6, 8),kexp 为2.7–3.0 × 107 M⁻1·s⁻1,不同全氟碳链长度的速率常数类似(图2a)。同样的,含甲基的n:2 FTCA(n = 6, 8)和含乙基的n:3 FTCA(n = 3, 5, 6, 7),kexp 分别为1.12−1.24× 107 M−1·s−1和1.20−1.57 × 108 M−1·s−1,进一步表明全氟碳链长度对氢提取反应活性没有显著影响。FTMePA与3:3 FTCA的速率相当,说明支链结构也不影响氢提取反应活性。然而,极性头基(磺酸根或羧酸根)及甲基/乙基取代的差异可导致kexp相差高达一个数量级,这可能与C−H键能、空间位阻及氢键作用有关,需在分子层面深入解析其引起速率差异的内在机制。
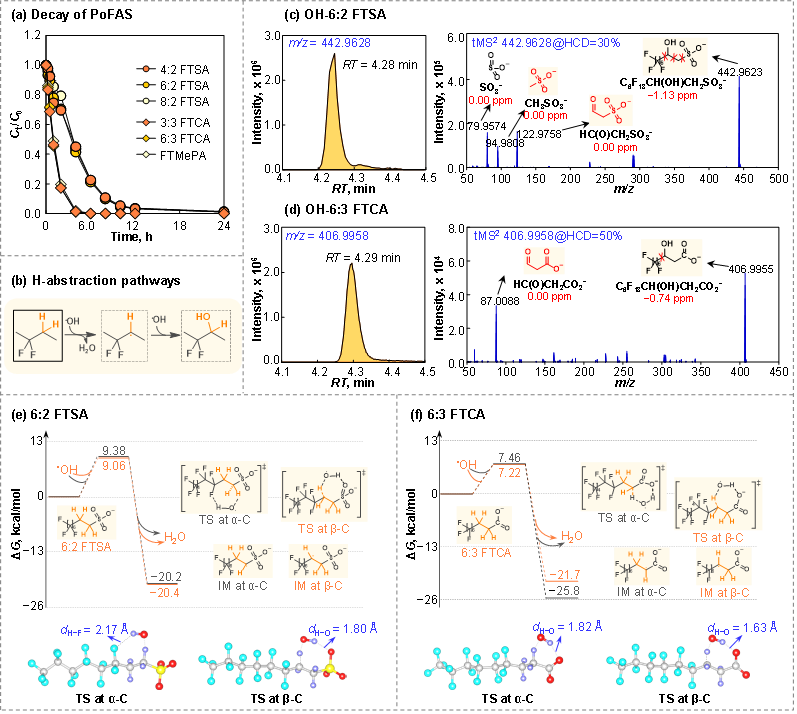
图2. 含甲基/乙基的PoFAS与•OH的反应活性及产物鉴定
对于含乙基的PoFAS,氢提取反应可发生在α-C或β-C位,生成相应的烷基自由基(−•CH−),随后与•OH反应生成羟基取代产物(HO-PFAS),并可通过质谱检测(图2b)。结果显示,HO-FTMePA存在两个保留时间不同的色谱峰,分别对应α-和β-位羟基取代产物,其二级质谱(tMS2)分别检测到α-位特征碎片离子HC(O)CO2− (72.9931, 0.00 ppm)和β-位特征碎片HC(O)CH2CO2− (87.0088, 0.00 ppm)。与之不同,n:2 FTSA与n:3 FTCA仅观察到一个色谱峰,其二级质谱仅检出β-位羟基取代的特征碎片,如CH3SO3− (94.9808, 0.00 ppm)、HC(O)CH2SO3−(122.9758, 0.00 ppm)和HC(O)CH2CO2−(87.0088, 0.00 ppm)(图2c、2d),表明β-C为其主要反应位点。进一步采用过渡态理论计算验证,结果显示β-C位氢提取反应的活化自由能(ΔG‡ = 7.22 kcal/mol,6:3 FTCA)略低于α-C(7.46 kcal/mol)位,同时因极性头基差异引起的空间位阻效应及过渡态中氢键的稳定性共同调控氢提取反应活性(图2e、2f)。反应动力学、转化产物与理论计算的一致性表明,过渡态理论能够有效捕捉并解释不同PoFAS结构所引起的反应活性差异。
对于其他类型的PoFAS,其反应活性也受结构特征显著影响。含单一C−H键的H-PoFAS,其氢提取反应活性较含甲基/乙基的PoFAS低了3个数量级,在UV/H2O2体系反应4小时后的转化率不足4%,相应的ΔG‡也较高、为10.7–13.0 kcal/mol。相比之下,含双键的烯烃类PoFAS,主要通过•OH加成机制反应,其速率远高于氢提取路径,相应的ΔG‡也较低、为5.58−10.5 kcal/mol。磺酰胺类PoFAS则主要通过其极性头基上的烃类结构发生多位点氢提取反应。过渡态理论计算表明,具有强氢键作用及环状过渡态构型的位点ΔG‡较低,是最可能的初始反应位点(图3)。例如,N-EtFOSAA中•OH倾向于攻击胺基上的乙基形成八元环过渡态,该结构通过与羧酸根形成氢键、并受磺酸基的电子吸引效应而稳定,其ΔG‡为3.79 kcal/mol;类似地,N-MeFOSAA中,最有可能的反应位点是胺基上的甲基,ΔG‡为4.35 kcal/mol。然而,由于反应生成的自由基(−•CH−)反应活性高、寿命短,难以在实验中直接捕捉和验证这些PoFAS的优先反应位点。
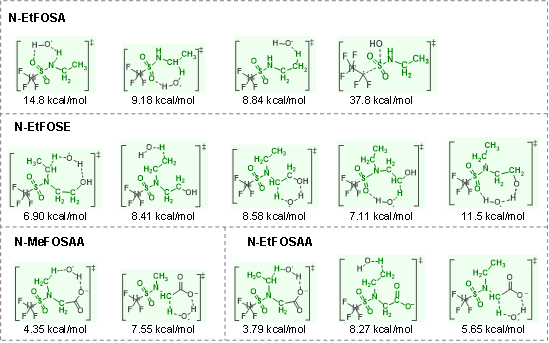
图3. 磺酰胺类PoFAS与•OH反应的过渡态结构及相应的活化自由能
对28种PoFAS进行理论计算显示,其理论速率常数(kTS)为1.91 × 103至7.32 × 109 M⁻1·s⁻1,与实验测定值(kexp)相差两个数量级以内。值得注意的是,kTS考虑了反应物在水相中的扩散效应。由于所研究的计算体系较大,当前理论值与实验值之间的差异处于可接受范围。kTS与kexp之间呈现显著的线性相关关系(r = 0.889,p < 0.01),表明基于过渡态理论的计算结果能够有效反映不同PoFAS与•OH反应活性的整体趋势(图4a)。需要指出的是,反应机制的类型对kTS与kexp的一致性具有较大影响。其中,含甲基/乙基的PoFAS相关性最高(r = 0.968,p < 0.01),而逐步纳入H-PoFAS、含双键(加成反应)及磺酰胺(头基氢提取反应)的PoFAS后,整体相关性逐步降低(图4b)。线性拟合结果显示,kTS可解释kexp变异性的79%(R2 = 0.790,p < 0.01)。进一步比较预测值与实测值发现,71%的理论预测值与实验值相差在1倍以内,92.9%的预测值偏差在5倍以内。这些结果表明,基于理论计算的速率常数可用于评估新型PoFAS与•OH的反应活性。特别对于缺乏标准品的PoFAS结构,该方法在不依赖实验动力学测定的前提下,为其潜在反应活性提供可靠预测。
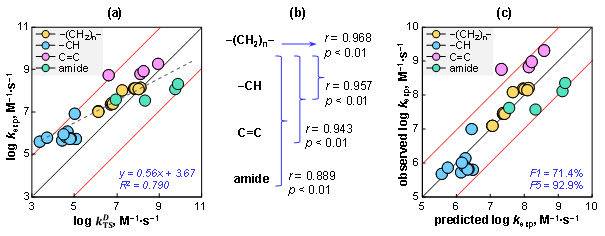
图4. PoFAS与•OH反应速率常数的理论值与实测值对比
PoFAS的非全氟基团不仅调控其与•OH的反应活性,也显著影响其反应路径与产物类型(图5)。PoFAS的转化产物可分为两类,一类是不能继续与•OH反应的稳定终产物(terminal products),主要包括全氟羧酸(PFCA)、全氟二元羧酸(PFdiCA)和全氟磺酰胺(FOSA);另一类是能与•OH继续反应的中间产物(intermediate),如含氧或羟基取代的化合物。
对于含甲基/乙基和双键的PoFAS,•OH通过氢提取或双键加成反应生成烷基自由基(−•CH−)。该自由基经•OH加和、氢提取、O2加成、HO2•消除等一系列反应,形成关键的酮基自由基 (−CO•(OH)−),进而引发C−C键断裂,生成全氟烷基自由基(CnF2n+1•)。其与•OH和O2反应,经HF消除和水解过程,最终生成短链PFCA(图5a、5b)。该反应路径在课题组前期研究中有详细阐述(Zhang et al., ES&T, 2021, 55 (24), 16655−16664)。H-PoFAS中C−H键在氢提取后转化为羧酸根结构,最终生成PFdiCA或一端为羧酸根、一端为磺酸根的终产物,例如HOOCCF2SO3−(图5c)。而磺酰胺类PoFAS则在头基发生氢提取后,通过C−S键断裂形成全氟烷基自由基(CnF2n+1•),进而转化为短链PFCA。
终产物与中间产物的分布高度依赖•OH的稳态浓度。当•OH稳态浓度较低或供应不足时,PoFAS转化易停留在中间产物阶段;而当•OH充足条件下,中间产物可进一步反应,最终转化为PFCA或PFdiCA。然而,这些终产物和中间产物,均保留了母体PoFAS中大部分的氟原子,即•OH氧化对PoFAS的脱氟降解能力有限。值得注意的是,部分终产物或中间产物可能具有比母体更高的生态毒性,例如6:2 FTSA的氧化产物6:2 FTCA对水生生物的毒性更强。因此,PoFAS在环境中的转化不仅增加了产物多样性,也可能提高其潜在环境风险。
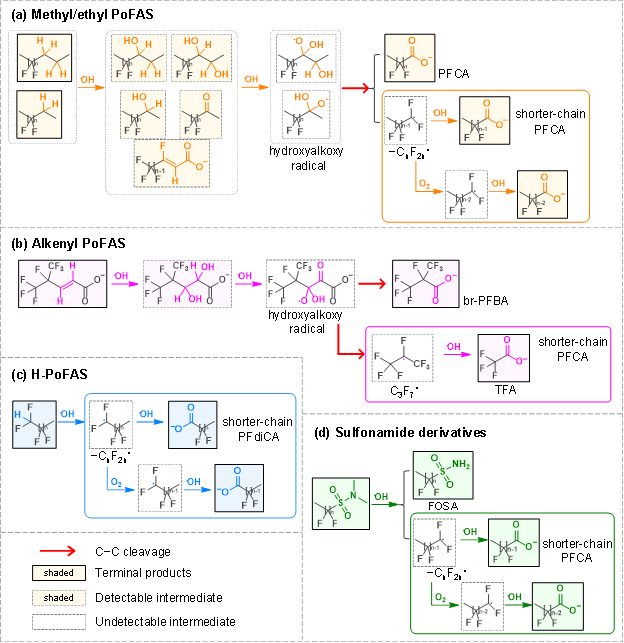
图5. •OH转化不同PoFAS的反应路径与产物特征
本研究系统阐释了•OH将多种非全氟基团转化为持久性更强的全氟羧酸类化合物的反应机制,证实了•OH介导的氧化过程是PoFAS在环境中的重要转化途径。过渡态理论有效解释了PoFAS结构对反应活性的调控机制,理论计算所得的速率常数与实验测定值在趋势上高度一致。通过解析过渡态结构,明晰了反应位点差异、极性头基、及空间位阻效应对反应活性的影响机制,深化了PoFAS在•OH体系中氧化行为的认知。研究中鉴定的终产物与中间产物扩展了环境样品中PFAS可疑筛查范围库。但是,PoFAS的转化程度和产物分布高度依赖•OH浓度及可能影响其浓度的水体基质成分,因此有必要在更接近实际环境条件下开展相关转化研究,以准确评估其环境行为。需要强调的是,PoFAS母体化合物的转化并不等同于环境风险的消除。为彻底解决PoFAS的污染危害,仍需发展能够实现PFAS全脱氟的降解技术体系。
本研究得到了国家自然科学基金委、浙江省自然科学基金委和西湖大学未来产业研究中心的资助。